Sickle Cell Disease in Kenya: Prevalence, Newborn Screening, Treatment Access and the Lake and Coastal Burden
Sickle Cell Disease in Kenya: Prevalence, Newborn Screening, Treatment Access and the Lake and Coastal Burden
Sickle cell disease (SCD) remains one of Kenya's most underappreciated public health challenges. Although the inherited blood disorder has been documented in East Africa for more than a century, the country only published a national newborn screening policy in 2020, and care for affected children continues to lag the disease burden. Approximately 14,000 Kenyan babies are born each year with sickle cell disease, and the prevalence is concentrated in well-defined geographies that reflect the historical pressure of malaria on the genome. This article maps the prevalence pattern across counties, examines treatment pathways from primary care to tertiary centres, considers the role of newborn screening, and explores the financing and human resource constraints that shape access to hydroxyurea, transcranial Doppler screening, and the small but growing transplant pipeline.
The Genetic Basis and Why the Lake Region Carries the Heaviest Burden
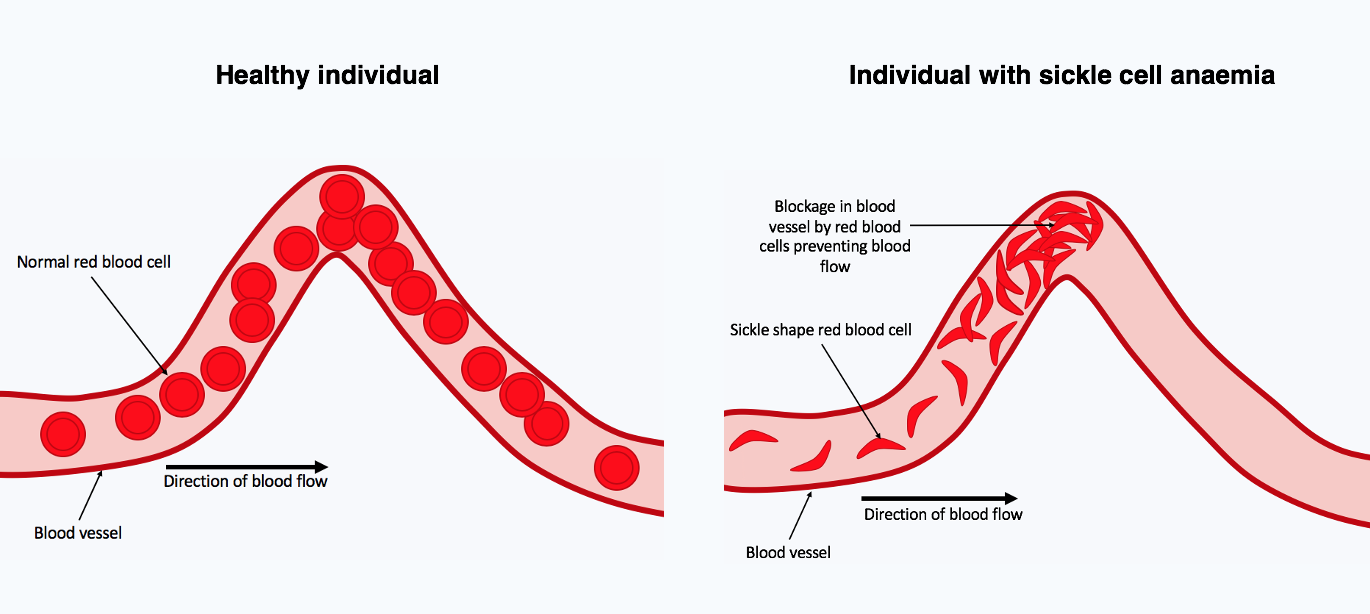
Sickle cell disease is caused by a single point mutation in the beta-globin gene that produces an abnormal haemoglobin known as HbS. When inherited from both parents the result is homozygous SCD, often presenting in the first year of life with painful crises, recurrent infections, splenic dysfunction and chronic anaemia. Heterozygous carriers, who inherit one HbS allele and one normal allele, have the sickle cell trait and remain largely asymptomatic. The trait confers partial protection against severe Plasmodium falciparum malaria, which explains why the gene has been positively selected in populations of the African Great Lakes and West Africa.
In Kenya the highest carrier frequencies are found in the lake counties of Homa Bay, Kisumu, Migori and Siaya, in the coastal counties of Kilifi, Mombasa and Kwale, and to a lesser extent in parts of Western Kenya and the lower Tana basin. Research published in PLOS One reports a 4.5 percent prevalence of sickle cell disease at birth in Western Kenya alongside an 18 percent prevalence of sickle cell trait, while infant cohorts in Homa Bay have recorded a disease prevalence as high as 9.6 percent. Counties of the Central highlands, Rift Valley pastoralist communities and arid northern districts carry a much lower load because the historical malaria pressure has been weaker.
Clinical Presentation and the Childhood Care Pathway
The clinical picture of SCD in Kenya typically declares itself between the third and twelfth month of life, when foetal haemoglobin levels fall and the proportion of HbS in the red cells rises. Parents commonly present with a child who has dactylitis, recurrent pneumonia, severe anaemia after a malarial episode, or unexplained jaundice. Without early diagnosis, mortality before the fifth birthday remains alarmingly high. Estimates from sub-Saharan cohorts suggest that more than half of affected children die before age five in settings without organised SCD care.
Once diagnosed, the standard of care in Kenya combines penicillin prophylaxis from birth through to at least age five, pneumococcal and Haemophilus influenzae type b vaccination, folic acid supplementation, parental counselling on hydration and fever recognition, malaria chemoprophylaxis where appropriate, and hydroxyurea therapy for moderate to severe phenotypes. Hydroxyurea, a cytotoxic drug originally developed for oncology indications, increases foetal haemoglobin production and reduces the frequency of vaso-occlusive crises, acute chest syndrome and transfusion requirements. The drug is now on the Kenya Essential Medicines List, although uninterrupted supply at county facilities remains uneven.
Comprehensive Care Centres and Tertiary Referrals
Kenya's principal sickle cell comprehensive care programmes are anchored at Moi Teaching and Referral Hospital in Eldoret, Kenyatta National Hospital in Nairobi, the Coast General Teaching and Referral Hospital in Mombasa, and the Jaramogi Oginga Odinga Teaching and Referral Hospital in Kisumu. Moi Teaching and Referral Hospital has the longest-running paediatric haematology service in Western Kenya, supported by partnerships with international academic centres and dedicated outpatient days that handle thousands of children annually. The Annals of Global Health reports that the Western Kenya programme has grown over more than a decade into a referral hub for Nyanza and Western counties.
Patients in level four and five county hospitals are often referred to these regional centres for the work-up of complications such as stroke, priapism, leg ulcers, avascular necrosis of the hip, retinopathy and renal involvement. Transcranial Doppler ultrasound, used to screen children at risk of overt stroke, is available in only a handful of facilities. Chronic transfusion programmes for stroke prevention remain limited by the safety and supply of blood. The Kenya National Blood Transfusion Service works with hospital transfusion committees to support paediatric haematology, but national blood collection remains below the World Health Organization recommendation of ten donations per thousand population per year.
The Newborn Screening Policy and the Implementation Gap
In 2020 the Ministry of Health published the national policy for newborn screening of sickle cell disease in facilities at levels two through six. The policy recommends universal screening in priority counties using high performance liquid chromatography or isoelectric focusing, with rapid point-of-care testing serving as a triage tool in lower-level facilities. Children who screen positive are to be enrolled into a comprehensive care register, started on penicillin prophylaxis and linked to vaccination, family education and clinic follow-up.
Implementation has been patchy. Homa Bay County, despite carrying among the highest prevalence in the country, has not yet rolled out routine newborn screening for SCD in its maternity wards. Sample logistics, reagent supply, laboratory technologist shortages and the absence of an integrated paediatric haematology cadre at primary facilities slow the cascade from screening to enrolment. Where screening exists, however, the acceptance rate among postnatal mothers is high. A PLOS One study from Western Kenya documented near-universal acceptability of newborn screening when mothers received a brief, culturally appropriate explanation alongside routine immunisation counselling. The implication is that household demand for screening is not the binding constraint, but rather the supply-side capacity of laboratories and clinics.
Diagnostics, Confirmation and the Role of County Laboratories
Point-of-care tests such as Sickle SCAN and HemoTypeSC have allowed Kenyan health facilities to screen newborns within minutes using a heel-prick blood sample. Positive results are then confirmed by a reference laboratory using high performance liquid chromatography or capillary electrophoresis. Reference capacity is concentrated in Nairobi, Eldoret and Kisumu, and most county facilities ship dried blood spot samples to these laboratories. Turnaround times of two to six weeks are common, which can delay the start of penicillin prophylaxis in remote sub-counties.
Genetic counselling for affected families, prenatal counselling for couples with known trait status, and pre-marital testing remain only sporadically available. Civic conversations about pre-marital genetic counselling have been gaining traction in faith-based forums and through patient advocacy groups, but the country lacks a coordinated counselling cadre. Strengthening laboratory networks, decentralising confirmation testing, and embedding pre-test counselling in antenatal care would all improve the screening cascade.
Financing, NHIF and the Out-of-Pocket Cost of Living with SCD
Living with SCD in Kenya is expensive for families. A typical year can include three to six hospital admissions for vaso-occlusive crises, multiple transfusions, hydroxyurea and folic acid prescriptions, antibiotics, vaccinations, school absences and travel costs to referral hospitals. The National Hospital Insurance Fund and the successor Social Health Insurance Fund cover inpatient admissions and selected outpatient packages, but families still bear significant out-of-pocket costs for medicines, laboratory tests outside hospital pharmacies and supportive nutrition. The country's transition to universal health coverage under the Ministry of Health is intended to expand chronic care benefits, including for inherited disorders, although implementation across counties remains uneven.
Patient advocacy organisations such as the Sickle Cell Federation of Kenya and county-level support groups in Kisumu, Nairobi and Mombasa have campaigned for the inclusion of hydroxyurea, transcranial Doppler screening and chronic transfusion packages in the benefits offered to chronic SCD patients. They have also pushed for the recognition of SCD as a disability under the Persons with Disabilities Act for those with severe phenotypes, in order to access educational and employment protections.
Hydroxyurea Scale-Up, Stem Cell Transplant and Emerging Therapies
Hydroxyurea is the single most cost-effective therapy currently available for moderate to severe SCD. Studies from Uganda and Nigeria have demonstrated that fixed dose hydroxyurea reduces hospital admissions, transfusion needs and mortality in African paediatric cohorts. Kenya has begun to expand access through hub-and-spoke prescribing, but the medicine still requires careful baseline laboratory work-up and regular monitoring, which presupposes functioning haematology laboratories at the spoke clinics.
Stem cell transplantation, the only currently approved cure for SCD, remains out of reach for most Kenyan families. A small number of patients have travelled abroad for matched sibling donor transplants in India, the United Kingdom and the United States. Domestic capacity is being developed at Kenyatta University Teaching, Referral and Research Hospital and at private oncology centres, but routine transplantation for SCD will likely require a decade of investment in tissue typing, donor registries and conditioning regimens. Newer gene therapies that re-engineer haematopoietic stem cells to produce foetal haemoglobin have shown promise internationally but remain prohibitively expensive at present.
Schools, Workplaces and the Social Dimension of SCD
Beyond medicine, the social dimension of SCD shapes outcomes profoundly. Children with SCD face stigma in schools, where teachers may interpret repeated absences as truancy. Painful crises during examinations, restricted participation in physical education and the appearance of jaundice can lead to bullying and social isolation. The Kenya Institute of Curriculum Development and the Kenya National Examinations Council have begun to incorporate accommodations for chronic illness, including extra time and rest breaks for sickle cell patients during national examinations, although implementation depends on parents securing medical reports in advance.
In workplaces, adults with SCD often face discrimination during recruitment and have to manage frequent sick leave. Public-sector employers are bound by the Employment Act and Persons with Disabilities Act, but private-sector enforcement remains inconsistent. Patient advocacy groups have called for clearer guidance from the Federation of Kenya Employers on reasonable accommodation for inherited blood disorders.
Research, Diaspora Engagement and the Way Forward
Kenyan researchers at the KEMRI Wellcome Trust Research Programme in Kilifi, the academic medical centres in Eldoret and Nairobi, and the regional health agencies have contributed important epidemiological, clinical and laboratory work on SCD over the past two decades. Diaspora professionals, including haematologists, transplant physicians and laboratory scientists, have supported training, mentorship and equipment donations. Diaspora-led foundations have funded screening kits, hydroxyurea procurement and patient support groups, and several Kenyan paediatric haematologists trained abroad have returned to lead programmes at home.
To close the gap between disease burden and care, the country needs sustained investment in newborn screening laboratories, decentralised hydroxyurea prescribing, predictable medicine supply chains, paediatric haematology training, blood transfusion capacity, and integrated comprehensive care centres at every regional referral hospital. With an estimated 14,000 affected newborns each year, even modest improvements in the screening, prophylaxis and chronic care cascade would translate into thousands of childhood deaths averted and a substantial reduction in family financial catastrophe.
Conclusion
Sickle cell disease in Kenya is no longer an invisible condition. The 2020 newborn screening policy, the expansion of hydroxyurea access, and the steady accumulation of comprehensive care programmes at major referral hospitals represent real progress. The remaining challenge is to translate national policy into county-level delivery, especially in Homa Bay, Kisumu, Kilifi, Mombasa and the other high-burden counties where most affected children are born. Building a national programme that screens at birth, treats with hydroxyurea, prevents stroke through transcranial Doppler, and supports families through schools and workplaces would mark a transformative step in Kenya's chronic disease agenda.
More Articles

The Multiparty Era Reforms: Saba Saba, the Repeal of Section 2A and Kenya's Return to Political Pluralism
Jul 02, 2026
Eye Care in Kenya: Cataract, Trachoma and the Long Road to Universal Vision Health
Jul 02, 2026

Shaba National Reserve: Joy Adamson's Last Wilderness and Isiolo's Semi-Desert Safari Gem
Jul 02, 2026

The Rendille People of Northern Kenya: Camel Pastoralism, Clan Traditions and a Culture Under Pressure
Jul 02, 2026

Ukulima Sacco: From Ministry of Agriculture Staff Society to a National Cooperative Open to All Kenyans
Jul 02, 2026